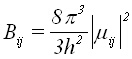 (1)
(1)
Интерес за химика представляват електронните спектри на многоатомните молекули. Електронната спектроскопия е първият молекулен спектрален метод, който е възникнал във връзка с потребността от изследване цветността на химичните съединения. Първата теория, въведена за обяснение цветността на химичните съединения е хромофорно-ауксохромната теория.Електронна спектроскопия
Част 2
Електронни спектри и структура на многоатомни молекули - увод
1. Хромофорно-ауксохромна теория
В 1876 г О. Вит изказва предположение, че цветността на едно химично съединение се дължи на наличието в него на атомни групировки, в които има ненаситени връзки. Такива групи (-N=N-, -C=O, -NO2, -NO и др.), които поглъщат определени дължини на вълната независимо от вида на съединението, в което присъствуват, той нарекъл хромофори.
Съществуват групи (-OH, -NH2, -SH и др.) или атоми - носители на свободни електронни двойки, които не преизвикват поглъщане във видимата област, но присъствието им в дадено цветно съединение предизвиква усилване на цвета. Такива “базични заместители” са наречени ауксохроми. Възприето е повишаването на интензитета на оцветяването (по-високият интензитет на абсорбционните ивици) под влияние на даден ауксохром да се означава като хиперхромен ефект. Обратният ефект, на понижение на интензитета е известен като хипохромен ефект.
Наред с въздействието върху интензитета ауксохромните групи променят и положението на абсорбционните максимуми (вж.по-долу). Преместването на максимума към по-големите дължини на вълната (червено отместване) се означава като батохромен ефект. Промяната на положението на ивицата към по-късите вълни (към виолетовата част на спектъра) се означава като хипсохромен ефект.
Понятието за хромофор се променя с разширяване възможностите за наблюдаване на електронни спектри извън видимата част на спектъра. Понастоящем като хромофор се разглежда всяка връзка, електронните преходи в която се извършват при незначително участие на електрони от други връзки и атоми в тази молекула. Тези характерни абсорбции лежат в основата на изследване на електронната структура на органичните съединения.
След хромофорно-ауксохромната теория са
предлагани много други теории, за обяснаване цветността на органичните
съединения, докато се достигне до квантовата теория. Взаимно допълвайки
се, експерименталната спектроскопия и квантовата теория са се развивали
дълги години заедно. Особено популярен сред химиците и плодотворен е споменатият
в предишната лекция метод
МО-ЛКАО. С негова помощ, използувайки експериментални данни от електронната
спектроскопия, се пресмятат редица молекулни параметри като: енергия на
преходите, йонизационен потенциал, диполни моменти, поляризуемост и др.
През последното десетилетие, успоредно с нарастване на изчислителната мощност
на персоналните компютри, в квантовата химия все по-масово се прилага неемпиричния
метод ab initio, с използуването на различни базиси (набори
от различни атомни вълнови функции).
2. Подборни правила и интензитет
Вероятността за абсорбционен преход се дава с уравнение (1), а интензитетът на съответствуващата ивица - с уравнение (2) - това на практика са три уравнения, понеже M е вектор.
(1)
където mi,j е така нареченият диполен момент на прехода, а h е Планковата константа, h = 6.62x10-34 J s.
От своя страна диполният момент на прехода може да се изчисли по уравнение (2)
![]() (2)
(2)
където Yi и Yj са вълновите функции на двете състояния, i и j, между които се извършва електронният преход, а M е векторен оператор, не зависещ от спиновите координати и имащ три реални координати, MX, MY и MZ.
Тъй като за многоатомните молекули точния вид на интеграла в уравнение (2) е неизвестен, информация за вида на вълновите функции Yi и Yj и на компонентите MK на диполния момент (MX, MY и MZ) се получава на основата на техните симетрични свойства. Такова разглеждане се извършва с теорията на групите, като правилата за подбор се получават от таблиците за характерите.
Интегралът в (2) ще бъде различен от нула само в случаите, когато произведението от подинтегралните функции се преобразува като напълно симетричино. Това условие се изпълнява ако произведението от характерите на Yi, Yj и MK е равно на едно пълносиметрично представяне. В случая на абсорбционни спектри на стабилни многоатомни молекули, основното състояние Yi е винаги пълносиметрично (A1 или A1g). Тогава преходът ще се извършва само на тези възбудени състояния Yj, за които прякото произведение от характерите на Yj и MK е равно на характерите на пълносиметричното представяне (диполният момент M е вектор и при молекули, притежаващи център на симетрия той се преобразува антисиметрично).
На основата на изложеното се разграничават три основни типа забрани за електронните преходи:
1. Забрана свързана с мултиплетността на състоянията: по време на прехода е забранена промяна на сумарния спин S, съответно на мултиплетността M, M = 2S+1.
Причина за това е обстоятелството, че понеже M е оператор, който не зависи от спиновите координати, то уравнение (2) може да се представи като уравнение (3), където интегралът е разделен на две части - при първия се интегрира по пространствените променливи на електрона, а във втория - по спиновите променливи. Ако имаме промяна на мултиплетността, например при преход от синглетно на триплетно състояние, то електронът ще си променя спина. Тогава в подинтегралната фунция на втория интеграл спиновите функции са a и b и те са ортогонални една на друга и следователно интегралът ще бъде нула, от което следва, че и моментът на прехода като цяло ще бъде равен на нула.
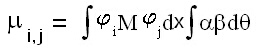 (3)
(3)
2. Преходи между молекулни орбитали, които не се припокриват пространствено или се припокриват съвсем слабо, са забранени. Това правило забранява преходите между s-oрбитали и p-oрбитали, както и преходи между свободни електронни двойки и s- и p--oрбитали, а разрешава преходите s а s* и p а p*.
3. Забрана по симетрия: преходи между орбитали с една и съща симетрия са забранени. За молекули притежаващи център на симетрия са забранени преходите jigаjjg и jiuаjju, т.е. ако прегледаме формата на молекулните орбитали от предишната лекция очевидно са забранени преходите sgаp*g и puаs*u,
Причина за това е обстоятелството, че всички компоненти MK на оператора на диполнития момент M на молекули от този вид са нечетни. Следователно интегралът в уравнение (2) ще бъде четен само ако М се умножи по една нечетна величина, което изисква вълновата функция на основното състояние Yi да бъде от различен симетричен клас от функцията Yj на възбуденото електронно състояние, т.е. за молекули с център на симетрия двете вълнови функции трябва да имат различин g-u характери.
Интегралът в (2) е едноелектронен, което предполага по условие, че възбуждането на повече от един електрон едновременно е забранено. Трябва да се отбележи обаче, че ако се отчете конфигурационното взаимодействие двуелектронното възбуждане може да се окаже разрешено.
Независимо от изброените забрани, за болшинството
от забранените преходи се наблюдават съответните ивици, но с нисък интензитет.
Най-силна е забраната по спин. От практическа гледна точка представлява
интерес класифицирането на даден преход като разрешен или забранен въз
основа на интензитета на ивицата. Разрешените преходи имат ивици с моларна
абсорбируемост e
> 104 [l mol-1 cm-1], а забранените -
e
< 103 [l mol-1 cm-1]
3. Видове електронни преходи
От направените по-горе разглеждания става ясно, че в молекулите на органичните химични съединения има три вида електрони:
- валентни електрони, с помощта на които се образуват молекулните връзки, имащи енергия, определена от съответните вълнови функции;
- свободни електронни двойки (n-електрони), които са локализирани на несвързващи атомни орбитали;
- вътрешни атомни електрони, които не участвуват в свързване и не могат да бъдат възбудени с електромагнитно лъчение от оптичната област на спектъра.
В зависимост от вида на участвуващите при преходите електрони, енергетичните преходи в органичните съединения се класифицират по следния начин:
1. N а V преходи. Към тези преходи се причислява възбуждането на електрон от свързваща на антисвързваща молекулна орбитала.
Съгласно забраната по симетрия (и припокриване) са разрешени само преходи от вида при молекулите с център на симетрия sgаs*u и puа p*g или sаs* и p аp* при молекулите без център на симетрия.
s аs* преходите (между молекулни орбитали на прости връзки) са свързани с абсорбция на ултравиолетово лъчение от далечната ??-област. Те не могат да се наблюдават с широкоразпространените спектрометри, поради което съединенията, притежаващи прости връзки, могат да се използуват като разтворители.
Най-голям интерес за органичната химия представляват pа p* преходите. Когато съединението притежава спрегнати кратни връзки pаp, преходът се означава като К-преход. К-преходите се проявяват в близката УВ и във видимата област с висока моларна абсорбируемост. Както положението, така и интензитетът на ивиците на тези преходи се променя под влияние на ауксохромните заместители.
2. N а Q преходи. Това са преходи, свързани с възбуждане на електрон от несвързваща на антисвързваща молекулна орбитала: nаs* и n аp*. Такива преходи се наблюдават при съединения, имащи в молекулата си хетероатоми, носители на свободни електронни двойки. Ивиците се проявяват в близката УВ и видима област с нисък интензитет, поради това, че са забранени преходи.
3. N а R преходи. Преходът на електрони в тези случаи се извършва от свързваща молекулна орбитала на основното електронно състояние върху орбитала на молекулен йон. Спектърът се проявява в далечната ултравиолетова област и представлява серия от ивици, завършващи с непрекъсната абсорбция.
От изложеното следва, че по принцип в молекулите на органичните съединения са възможни следните електронни преходи: sаs*, pаp*, nаs* и n аp*. На фигура 4 е представена схематично енергетичната диаграма на тези преходи.
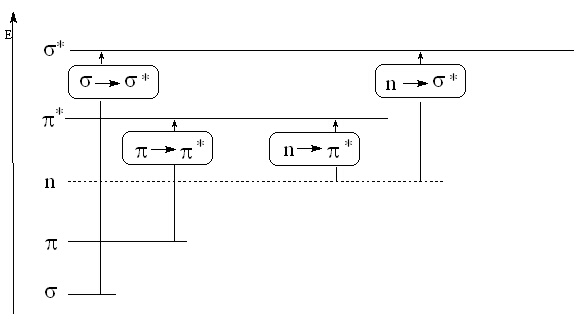
Фигура 4. Електронни преходи в молекулите на органичните съединения.
4. Пътища на дезактивация на електронното възбуждане. Времена на живот на възбудените състояния.
В досегашните разглеждания ние се спряхме по-подробно на абсорбционните и емисионни (оптични) преходи. Една по-пълна представа за процесите, настъпващи при взаимодействие на електромагнитното лъчение с веществото, дава диаграмата на Яблонски. На фигура 5 е представена една нейна модификация.
От основно състояние S0
системата преминава след поглъщане на квант енергия в по-висвисоко
енергетично състояние S1, S2,
…. Sn за около 10-15 s, където престоява около
10-8 s. Връщането в основно състояние се извършва по два принципно
различни
начина - оптически, т.е. чрез излъчване
на погълнатата енергия или безизлъчвателно (неоптически преход).
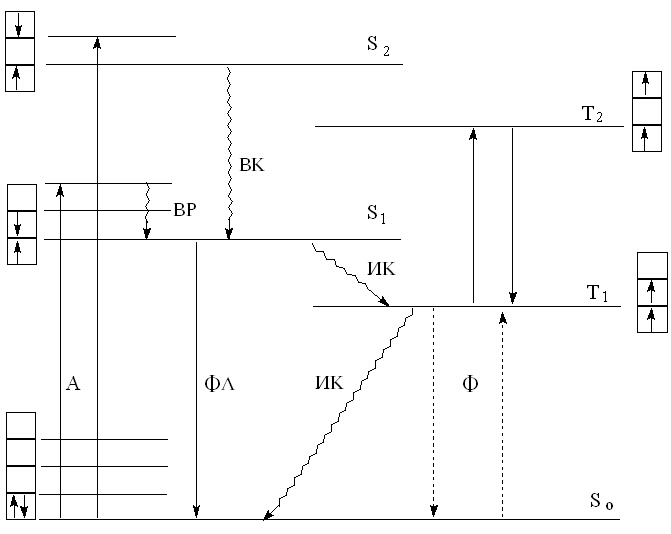
Фигура 5. Модифицирана диаграма
на Яблонски за видовете електронно-вибрационни преходи в органични молекули.
| Оптични:
А - абсорбция ФЛ - флуоресценция Ф - фосфоресценция |
Безизлъчвателни:
ВК - вътрешна конверсия ИК - интеркомбинация ВР - вибрационна релаксация |
Връщането в основно състояние S0 чрез излъчване се извършва най-често от S1, а много рядко от по-горни нива (Sn). Такъв преход се нарича флуоресценция (ФЛ) - фигура 5. При това, колкото е по-голяма вероятността за преход на електрон във възбудено състояние, толкова по-голяма е вероятността за връщане в основно състояние. От тук следва, че колкото по-интензивно поглъща едно съединение при определена честота, толкова по-кратък е престоя му във възбудено състояние.
Безизлъчвателното дезактивиране до S1
или S0 се нарича вътрешна конверсия (ВК).
Когато вътрешната конверсия се извършва между няколко вибрационни състояния
на едно електронно състояние, тя се нарича вибрационна релаксация (ВР).
Скоростната константа на този тип дезактивация е около 1012
s-1. Безизлъчвателният път на дезактивация е доминиращ при
вибрационните преходи.
Разгледаните дотук начини за превръщане на погълнатата енергия се извършват между различни електронни състояния, притежаващи една и съща спинова мултиплетност.
Превръщането на погълнатата от молекулата енергия може да бъде съпроводено с промяна на спина на възбудения електрон, т.е. до преминаване в т.н. триплетно състояние Т. Безизлъчвателен процес между две електронни състояния, свързан с промяна на спина на електрона, се нарича интеркомбинация (ИК). Връщането на системата в основно синглетно състояние S0 може да стане отново безизлъчвателно (интеркомбинация) или оптически, чрез излъчване на фотон. В последния случай процесът се нарича фосфоресценция (Ф). Времето на живот в това състояние е много дълго (10-2 - 104 s) поради това, че преходът триплет-синглет е забранен. Спиновата забрана се снема частично поради взаимодействие между спиновото и орбитално движение на електрона. Оптичният преход синглет-триплет е също забранен (преходът е даден с пунктир), докато преходите триплет-триплет са разрешени. Последното се използва в някои случаи за създаване на източници на лъчение. Например, молекулен водород или деутерий в условията на електричен разряд преминават от по-високо в по-ниско възбудено триплетно състояние, при което се излъчва ултравиолетово лъчение.
Оптичните преходи на дезактивация ФЛ и Ф се обединяват под общото наименование луминесценция (Л). Понеже при тези процеси част от погълнатата светлинна енергия се отдава на системата безизлъчвателно, превръщайки се в топлина, най-често по пътя на ВР, спектрите на ФЛ и Ф са отместени към по-големите дължини на вълната спрямо съответната абсорбционна ивица. Когато спектрите са заснети в неполярни разтворители, обикновенно се наблюдава фина структура на електронните ивици. Фината структура на абсорбционните ивици отразява състоянието на вибрационните нива на възбуденото електронно състояние S1, докато вибрационната структура на луминесцентните ивици характеризира вибрационните нива на основното електронно състояние S0. Между електронно-вибрационните спектри на А и Л понякога се наблюдава т..н. огледална симетрия.
Когато даден електронен преход се извършва не от нулево, а от някое възбудено вибрационно подниво на S0, съответните ивици се наричат “горещи” ивици. Те се проявяват в спектъра при дължина на вълната по-голяма от тази на 0 а 0 прехода.
Всички описани дотук процеси на дезактивация са спонтанни. В някои случаи е възможно и извършване на принудително излъчване. Този процес се осъществява при облъчване на дадена възбудена молекула с лъчение, честотата на което е равна на луминесцентната честота на молекулата. Връщането на молекулата в основно състояние в този случай се съпровожда с едновременно изпускане на два фотона - на луменисценция и индуциращия го. При това двата фотона са неразличими; те притежават еднаква честота, фаза и насоченост. Този процес се нарича стимулирано излъчване. Задължително условие за извършване на стимулирано излъчване е по-голямата заселеност на възбуденото, от тази на основното състояние (N0 < N1), т.н. инверсно заселване. Последното се постига по следния начин: поглъщайки светлинен квант, системата преминава от основно в някое от разрешените възбудени синглетни състояния. От там тя може да се върне в основно чрез ИК до някое по-ниско синглетно ниво, времето на живот в което е по-дълго. Тъй като скоростта на дезактивация на това състояние е много по-малка, отколкото скоростта на преминаване в по-горното възбудено ниво, броят молекули, заселващи по-ниското възбудено ниво бързо нараства и става по-голям от този на основното състояние. Инверсно заселване може да се постигне и за някои вибрационни или ротационни нива (в рамките на едно електронно, респективно вибрационно състояние), когато времето на живот в това междинно ниво е по-голямо от времето на живот в нивото, до което състемата достига след абсорбция на кванта. Такива системи могат да генерират стимулирано излъчване. Описаните процеси играят важна роля при създаване на лазерни лъчения.
Освен по описаните дотук начини, превръщането на погълнатата енергия може да се извърши също чрез химически (фотохимично разпадане на молекули) или чрез бимолекулярни фотофизически процеси.
Литература
Автор: Проф. дхн Георги Андреев